If you’ve recently watched NBC Nightly News you may have heard of the impending helium shortage (the topic for our next post), but there is another shortage, perhaps as equally impacting as the helium shortage, that most people have not heard about. That’s because this shortage happens all the time. It is the drug shortage. Where did all the drugs go? They didn’t go to Colorado as some might suspect with the recent legalization of marijuana. These are prescription, FDA-approved drugs that are on shortage, not recreational drugs normally run by drug cartels. But what’s even more interesting is that the drug shortage isn’t a normal shortage. As explained before, these shortages occur all the time, but only to a select number of prescription drugs, and these shortages are much more temporary compared to the helium or Plutonium-238 shortage. Nevertheless, that doesn’t mitigate the impact of this constant shortage. So why is there a drug shortage? As the FDA explains, these drug shortages are due to a number of reasons, most commonly manufacturing or quality problems. As we explore both of these factors, and examples of current drug shortages, you will see how both issues can affect a large population, not tomorrow or in 2025, but now.
To understand a little more about the underpinnings of this unknown shortage, you must first understand how competitive and difficult it is to bring a drug to market. The process often takes a couple of years and a couple billion dollars. The tests for safety are rigorous and lengthy and the paperwork fills dictionaries. It is no wonder why drugs are so expensive and why drug companies will do everything they can to make more money…
So what goes into this testing process regulated by the seemingly “devilish” FDA? Once a company has created a new drug, it can begin the FDA’s Drug Review Process. To start, there are preclinical animal testings. This phase is simply to identify whether the drug is lethal at its prescribed dosage. If it kills a mouse, you certainly do not want to give it to a human. Results must be shown to and reviewed by the FDA before human trials can begin. This transition to human clinical trials is marked by an Investigational New Drug (IND) Application. The first human trial is called Phase 1 where 20-80 healthy patients are given the drug simply to gauge the drug’s safety in humans and discern how the drug is metabolized and excreted. If it is determined that there isn’t an unreasonable toxicity Phase 2 trials begin. Phase 2 tests the effectiveness of the drug compared to placebos in treating a certain ailment. Typically a few dozen to 300 patients partake in Phase 2. If the drug is determined as effective, Phase 3 begins. Phase 3 are mass testings, often with a few thousand participants. This phase tests the drug’s interaction with other drugs and once again the drug is reviewed for its safety and effectiveness. The results are reviewed and finally all phases, trials and results are reviewed in a New Drug Application (NDA). Not so bad? The entire process can take up to 12 years, and drugs have a 1 in 5000 chance of making it through the process.
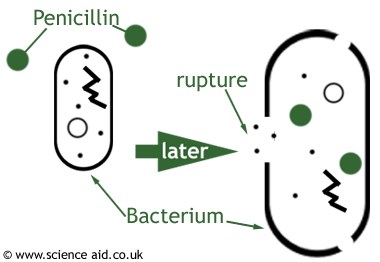
What does this have to do with the problem at hand? Well, besides bankrupting any small company, the FDA regulates drugs on a much lower frequency once a drug is brought to market with the thought that these drugs are safe. However, this means that your safety is not guaranteed. Often times a demand for a drug will increase, while just as frequently a manufacturing company may close down thereby decreasing supply. This is how a manufacturing and consumerism problem can lead to a shortage. But remember, the drug company just paid, on average, 4 billion dollars to bring that drug to market, there is no way a small manufacturing problem is going to stop the drug company from selling their drug. So what might a drug company do? Come up with a cheaper, more efficiently manufactured formula for the drug and begin manufacturing that. What’s to stop them? The FDA is not due for a visit in another year – hopefully the new formula won’t hurt anyone. And that’s how quality problems arise. A simple change in the formula harms a select, but noticeable, clientele and they report the problem to the FDA. That’s when the FDA’s visit to the drug company is unexpected and a lot sooner than their annual or biannual visit. The FDA will halt all production of the drug, and begin running testings and reviews of the drug once more. So as you can see the two factors – manufacturing and quality – are interlinked and sometimes a drug company will simply pick the lesser of two evils. This makes for a very harmful shortage not only when the formula of the drug is changed, but also in either scenario when production of the drug is slowed or halted and the patientneeding the drug cannot get it.
So what are some current drug shortages, what do the drugs do and who do the shortages affect? An interesting one with many applications is Dexamethasone Sodium Phosphate Injections.

 [1]
[1]
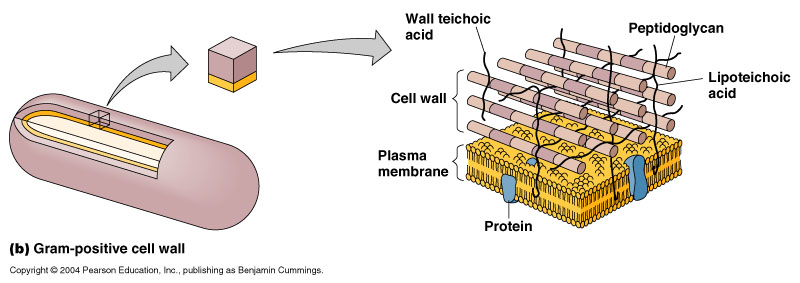
Dexamethasone is a potent glucocorticoid (the term glucocorticoid is interchangeable with corticoid or corticosteroid) and the injection of Dexamethasone Sodium Phosphate is used to treat allergic reactions, arthritis, blood diseases, breathing problems, eye diseases, intestinal disorders and more. It can also be used to diagnose an adrenal gland disorder (Cushing’s disease) since the adrenal gland is responsible for secreting corticoids. Dexamethasone and other corticoids are anti-inflammatory and is the reason why they can be used to treat ailments like allergic reactions, arthritis or skin disorders (such as bug bites). This is because dexamethasone binds to the glucocorticoid receptor – since they are complementary in structural shape [1] – which is responsible for suppressing the immune system (and heightening awareness). During times of stress, the adrenal gland releases corticoids, which bind to the glucocorticoid receptor. The body suppresses all non-essential functions (i.e. the immune system) and heightens all functions beneficial to survival (i.e. attention). This natural phenomena is used to stop inflammation, a natural immune response. It is also the reason why withdrawing from dexamethasone or any other corticoids can cause weakness and tiredness.
Because of its versatility, the dexamethasone sodium phosphate injection shortage affects many people who need a wide variety of treatments. There are 4 manufacturers of the dexamethasone sodium phosphate injection, but over half of the concentrations of the injection are unavailable from all suppliers. One concentration from a certain supplier may not recover until April, 2014 because of increased demand. Most of these backorders or unavailability began in January of 2013.
Calcium chloride injections are also in shortage, and while they aren’t as widely applicable as Dexamethasone Sodium Phosphate injections, their absence is just as life-threatening. Calcium chloride (CaCl2) is normally a crystal structure as depicted in the below two pictures.


Its crystal structure forms a rather unique cubic unit cell with a total of 2 Calcium atoms and 4 Chloride atoms – appropriately so since there should be twice as many Chloride atoms as there are Calcium atoms. 8 Calcium atoms reside in each corner of the unit cell, and since each contribute 1/8 to the unit cell, that is a total of 1 Calcium atom. Another Calcium atom resides in the center of the unit cell. What makes this unit cell so interesting though is the placement of the Chloride atoms make this cubic unit cell neither a face-centered unit cell nor a body-centered unit cell. 2 Chloride atoms reside completely within the unit cell, while 4 Chloride atoms reside on the faces, each contributing 1/2 to the unit cell which accounts for the 4 total Chloride atoms that are within the cubic unit cell.
As a solid, Calcium chloride is commonly used as brine for refrigeration plants, salt on roads to prevent ice, and desiccation, or the removal of water by chemical means. However as a liquid, the Calcium chloride injection is used for immediate treatment of hypocalcemic tetany, uncontrollable muscle contractions and spasms due to a lack of Calcium in the body, treatment for insect bites, aid for depression due to an overdose of Magnesium sulfate (which happens to be another drug currently on shortage – not because of an increase in overdoses, and the two shortages are not related) and to improve weak or ineffective myocardial  contractions when epinephrine (in injection form is also on shortage. Again, the two shortages are not related) has failed to due so (usually after open heart surgery). Calcium chloride injections are vital to both hypocalcemic tetany, since the cause of the illness is a lack of calcium, and recoveries from open heart surgeries. Again, while both diseases are rare and far less common than the diseases dexamethasone sodium phosphate injections treat, this shortage can have detrimental effects. The shortage began in December of 2012, and still has yet to recover since half the suppliers have either discontinued manufacturing calcium chloride injections, or were having trouble manufacturing enough for the increased demand.
contractions when epinephrine (in injection form is also on shortage. Again, the two shortages are not related) has failed to due so (usually after open heart surgery). Calcium chloride injections are vital to both hypocalcemic tetany, since the cause of the illness is a lack of calcium, and recoveries from open heart surgeries. Again, while both diseases are rare and far less common than the diseases dexamethasone sodium phosphate injections treat, this shortage can have detrimental effects. The shortage began in December of 2012, and still has yet to recover since half the suppliers have either discontinued manufacturing calcium chloride injections, or were having trouble manufacturing enough for the increased demand.
The final shortage worth touching upon is the copper injection shortage which is often delivered as cupric sulfate (Cu(II)SO4).

This interesting cubic unit cell is even more complex than that of calcium chloride. Again it is neither a simple cubic, face-centered cubic nor body-centered cubic unit cell but rather a combination of them all “plus more”. The copper ions reside in the corners of the unit cell, the edge of the unit cell, the face of the unit cell and in the center of the unit cell. A copper ion resides on each corner and since each contribute one eighth of an ion (8*1/8) that amounts to 1 ion. Next there are 4 edge cells which contribute one fourth of an ion (4*1/4) which totals another ion. Then there are 2 face centered ions which each contribute half an ion (2*1/2) which is another ion. Finally there resides one copper ion in the center of the unit cell. In total there are 4 copper ions. Further evaluation of the unit cell concludes in the fact that there are 4 sulfur ions and 16 oxygen ions within the unit cell, or otherwise 4 sulfate (SO4-2) ions. This makes sense since the ratio of copper to sulfur to oxygen ions is 1:1:4 and the ratio of copper ions to sulfate ions is 1:1.
Copper injections are used in IV therapies (Intravenous therapies) as a part of total parenteral nutrition (TPN). Copper is necessary, in small quantities, in a human diet and when a patient cannot eat, it is required that they receive their nutrients in another manner; IV therapies. IV therapies are used often in hospitals so this shortage may have a significant impact on hospital patients and is the reason why this may be the most urgent shortage mentioned in this blog. The shortage is not only life threatening to those who need it, but it also affects a large population. The shortage began in April of January and the sole manufacturer has halted production of the injection. The only signs of hope are Cupric Chloride injections, another form of copper injections which are expected to recover from their own shortage sometime this quarter.
Hopefully, as you can begin to see, the fairly unknown drug shortage, is a very serious problem. Luckily most shortages are due to manufacturing delays and not quality problems (perhaps drug companies are not as greedy as the public might believe), but the shortages which can last months, are still detrimental to those who need the drugs. As the problem continues unnoticed by the general public, the best we can hope for is a revamping of manufacturing of dozens of drugs on shortage to satiate the ever-increasing demand for drugs.



 Yellowstone National Park
Yellowstone National Park
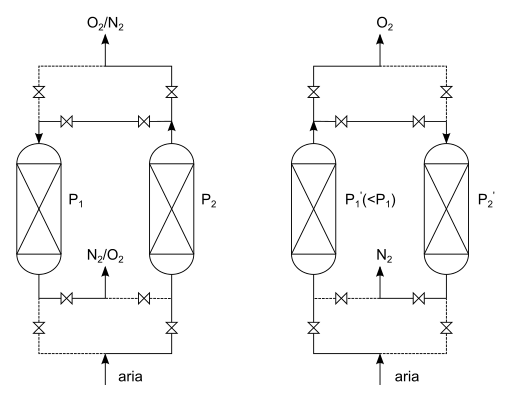 Pressure Swing Absorption Process
Pressure Swing Absorption Process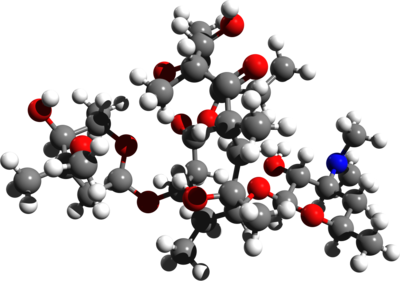
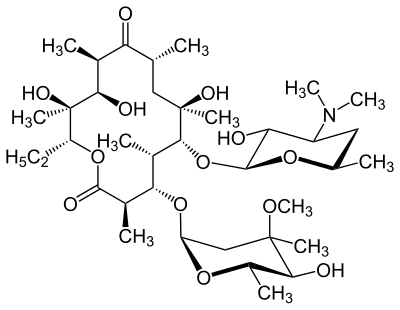

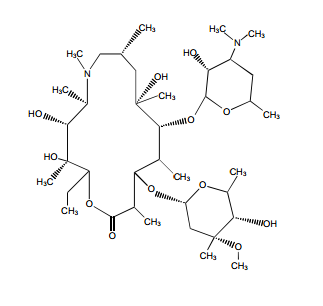
 tidepressanst that are currently on the market, representing a now aged class of treatments combating depression. Muscarinic, histaminergic and α1-adrenergic receptors are antagonized in the action of classical TCA drugs, leading to anticholinergic (rendering inactive the neurotransmitter acetylcholine), sedative, and cardiovascular effects. In vitro, fluoxetine unites with the aforesaid receptors in the brain tissue with less efficacy than TCA drugs. As identifiable through their names, these TCAs have a three-ring chemical structure. For example,
tidepressanst that are currently on the market, representing a now aged class of treatments combating depression. Muscarinic, histaminergic and α1-adrenergic receptors are antagonized in the action of classical TCA drugs, leading to anticholinergic (rendering inactive the neurotransmitter acetylcholine), sedative, and cardiovascular effects. In vitro, fluoxetine unites with the aforesaid receptors in the brain tissue with less efficacy than TCA drugs. As identifiable through their names, these TCAs have a three-ring chemical structure. For example,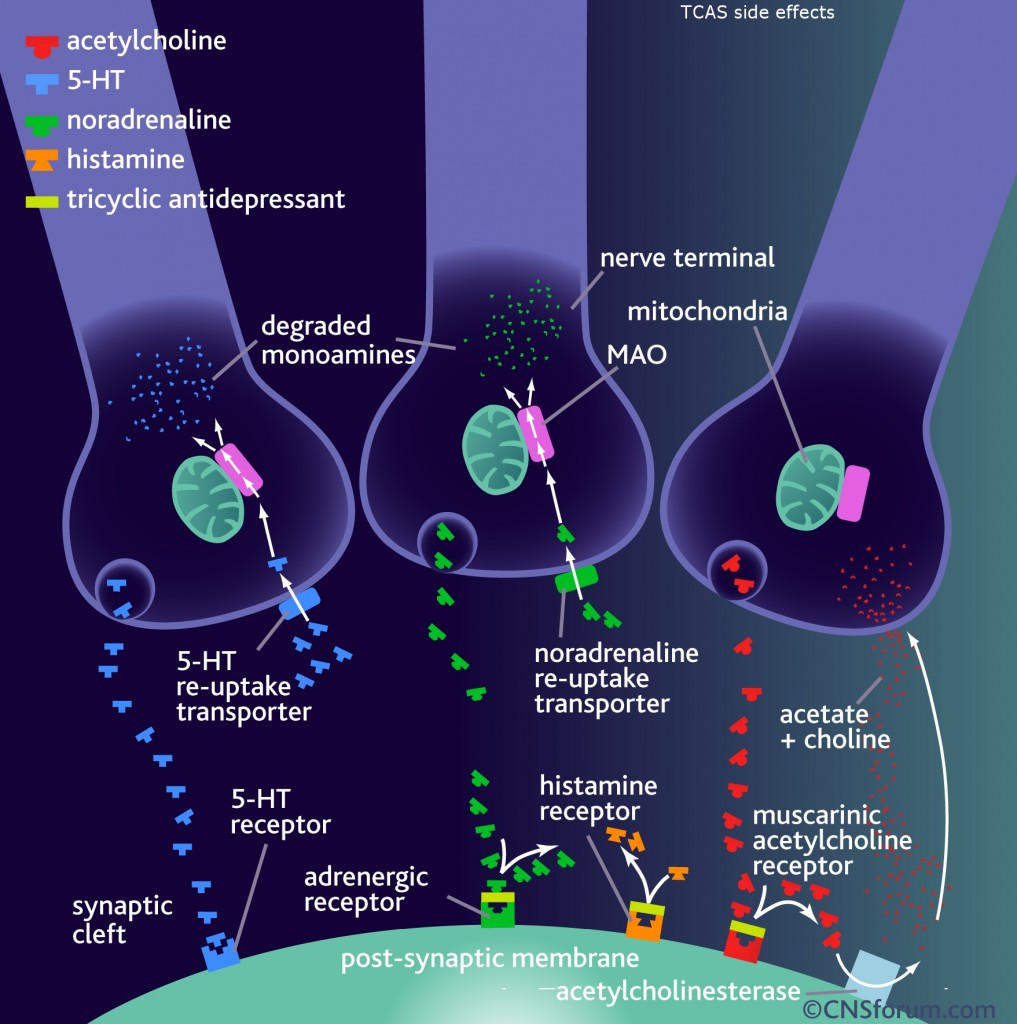 in
in 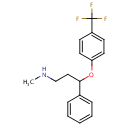 d by the location of an asymmetric carbon atom in the general structure. This is a feature to be noted due to its usages in inorganic, organic, physical, and biological chemistry. It is metabolized by CYP2D6 by the liver, characterized by its slow rate and a long half-life in the confines of the system. Slow aggregation leads to delay in the manifestation of meaningful effect. It is also an agonist for 5HT2C receptors, linking back to the first blog post on beta-agonists.
d by the location of an asymmetric carbon atom in the general structure. This is a feature to be noted due to its usages in inorganic, organic, physical, and biological chemistry. It is metabolized by CYP2D6 by the liver, characterized by its slow rate and a long half-life in the confines of the system. Slow aggregation leads to delay in the manifestation of meaningful effect. It is also an agonist for 5HT2C receptors, linking back to the first blog post on beta-agonists.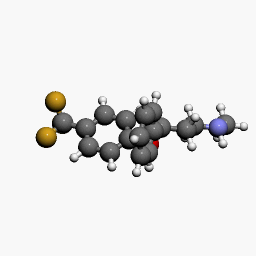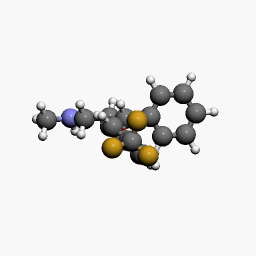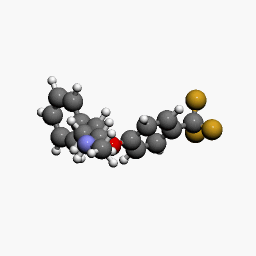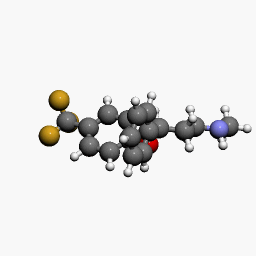








 Pictured here is a dopamine neuron releasing dopamine, depicted as yellow-green circles, to a nearby neuron. Dopamine is responsible for making humans feel “good.”
Pictured here is a dopamine neuron releasing dopamine, depicted as yellow-green circles, to a nearby neuron. Dopamine is responsible for making humans feel “good.” Pictured here is a woman who is suffering from opioid-induced hyperalgesia after long-term opioid use.
Pictured here is a woman who is suffering from opioid-induced hyperalgesia after long-term opioid use.




{kind=link}
{kind=link}
{kind=link}