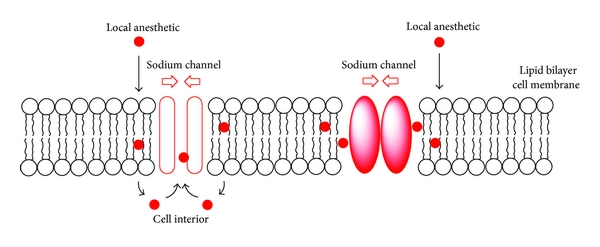
As seen in the previous post, local anesthetics generally work to prevent only a small area of the body from experiencing pain by inhibiting the flow of sodium ions (preventing action potential thus preventing nerve activity) through sodium channels embedded in the cell membrane of neurons. More specifically, the local anesthetic will bind to a receptor inside the sodium channels and antagonize it, therefore closing the sodium channels thus creating the halt in the influx of ions through the channels as seen in the diagram below.
.
Many local anesthetics commonly bind to the N-methyl-D-aspartate (NMDA) receptor (an image of how the anesthetic might bind to a receptor through the polar attractions between the receptor and anesthetics is shown here), such as the constituents of the Caine family: a category of local anesthetic compounds that share similar qualities (i.e. similar receptors and mechanism of actions) and end in the suffix “caine”. The following will consist of descriptions of three different local anesthetics, particularly from the Caine family, to demonstrate the functional and molecular diversity in the compounds of local anesthesia.
Cocaine:
Cocaine, otherwise known as benzoylmethylecgonine, can be used as a type of local anesthetic, but for the past several decades it has reached the headlines for different reasons. Cocaine was used historically as an eye and nose anesthetic, used to block nerve signals during surgery, but side effects of cocaine exposure during surgery include intense vasoconstriction and cardiovascular toxicity. It is a powerful nervous system stimulant, and above all, it is extremely addictive. Repeated use of the drug can cause strokes, cardiovascular disease, and several hundred other afflictions such as gingivitis, lupus, and an increased chance for heart attacks. Cocaine can be administered in many different ways, most commonly through insufflation, injection, and in the case of crack cocaine, inhalation. Cocaine is a controlled substance around the world due to its addictive properties and terrible side effects of constant use.
How To Use It:
Most users of pure cocaine are drug addicts, but cocaine hydrochloride is still used as a topical anesthetic. It is applied through the mouth, or the nose using a cotton swab to numb the area. It should not be used in the eye or injected, and rarely, addictive behavior will be expressed by the patient. Use the medication as specified by a healthcare professional, and do not use more frequently or longer than specified.
Molecular Structure:
Cocaine usually contains pure C17H21NO4 from the leaves of the coca plant.]
2D structure 3D structure
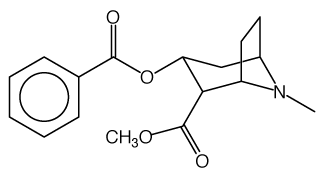
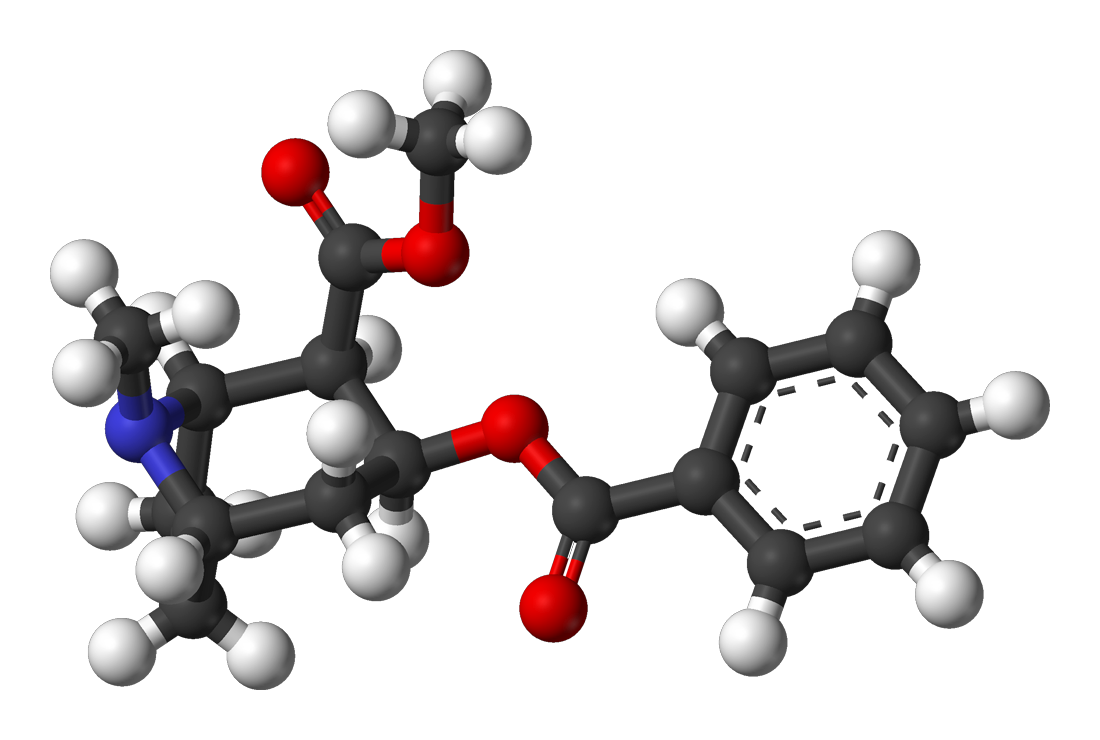
Properties:
- The molecular weight of cocaine is 303.35 g/mol.
- The molecular formula is C17H21NO4
- The systematic name is Methyl (1R,2R,3S,5S)-3-(benzoyloxy)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate
- Approximately 35.9 million Americans aged 12 and older have tried cocaine at least once in their lifetime, according to a national survey, and about 2.1 million Americans are regular users
Novocain (Procaine):
First synthesized in 1905, novocain (the trade name of procaine) is an ester-type local anesthetic that is able to induce a loss of sensation when injected, as opposed to oral intake which has been stated to wield therapeutic values. The first synthetic local anesthetics to be produced, novocain was primarily utilized for oral surgeries in dentistry however due to ester-type anesthetics having generally a high potential of causing allergic reactions, it eventually became obsolete and eventually replaced by a more effective anesthetic known as lidocaine. Ester-type anesthetics are more prone causing allergic reactions compared to Amide-type anesthetics because when they metabolize in the body, they form a compound known as para-aminobenzoic acid (PABA). PABA has a documented history of causing allergic reactions that range from urticaria to anaphylaxis. Generally, the adverse side effects of using novocain include heartburn, migraines, nausea, and can induce a serious condition known as systemic lupus erythematosus (SLE), therefore it is highly advised that intake is performed by a healthcare professional. However, novocain also retains the property and advantage of constricting blood vessels, reducing bleeding unlike many other local anesthetics.
How To Use It:
The common and primary method of intake of novocain for its anesthetic properties is through injection in solution state. However, if novocain is present in capsule or tablet form, oral ingestion can also performed though its properties and effects will be greatly mitigated and may induce therapeutic rather than anesthetic conditions. An informative video of how novocain is administered in oral surgeries of dentistry can be found below.
Molecular Structure:
Novocain contains pure C13H20N2O2.
2D structure 3D structure
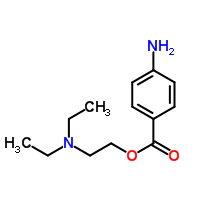
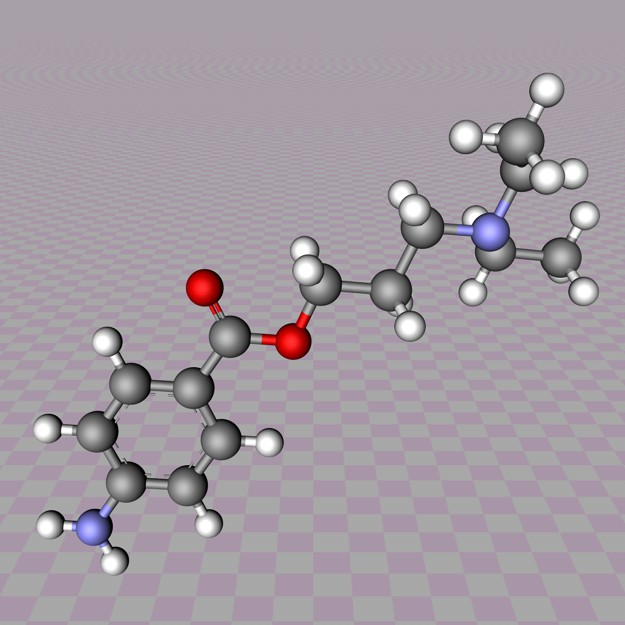
Properties:
- The molecular weight of novocain is 236.31 g/mol.
- The molecular formula is C13H20N2O2.
- The systematic name is 2-(diethylamino)ethyl 4-aminobenzoate
- The melting point of novocain is approximately 61 °C while its pKa value at 15 °C is 8.05
Tetracaine:
Tetracaine is a type of local anesthetic and it is used as a numbing medication. It is generally used for surface and spinal anesthesia and it works by blocking the nerve signals in your body. There most used type of tetracaine medication is cream and ointment. It’s primary use is to reduce pain or discomfort caused by minor skin irritations, cold sores or fever blisters, sunburn or other minor burns, insect bites or stings, and many other sources of minor pain on a surface of the body. The reason why this medication is given is to lessen the pain caused by the insertion of a medical instrument such as a scope or a tube. Although in most situations tetracaine is used on the skin, it can also be used on the eye. This eye medication is in the form of drops and it is used to decrease the feeling in your eyes right before going through surgery or perhaps a test or procedure involving the eyes.
How To Use It:
The eye drops medication should be issued by the clinic and after going through the procedure, the patient must refrain from touching his or her eye until the medication is no longer in effect and in some cases, an eye patch is required. The Tetracaine topical gel is applied by very small amounts only necessary to cover the area and should not be used more than four times a day unless the doctor specifies otherwise.
Molecular Structure:
Tetracaine contains more than 98 percent of .C15H24N2O2 calculated on the dried basis..
2D structure 3D structure
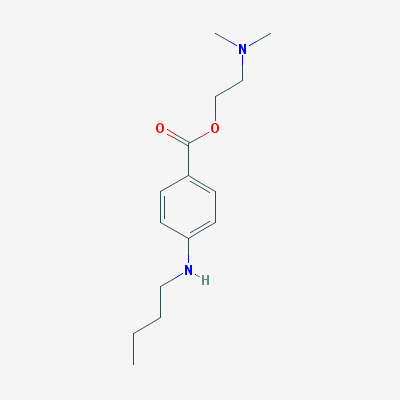
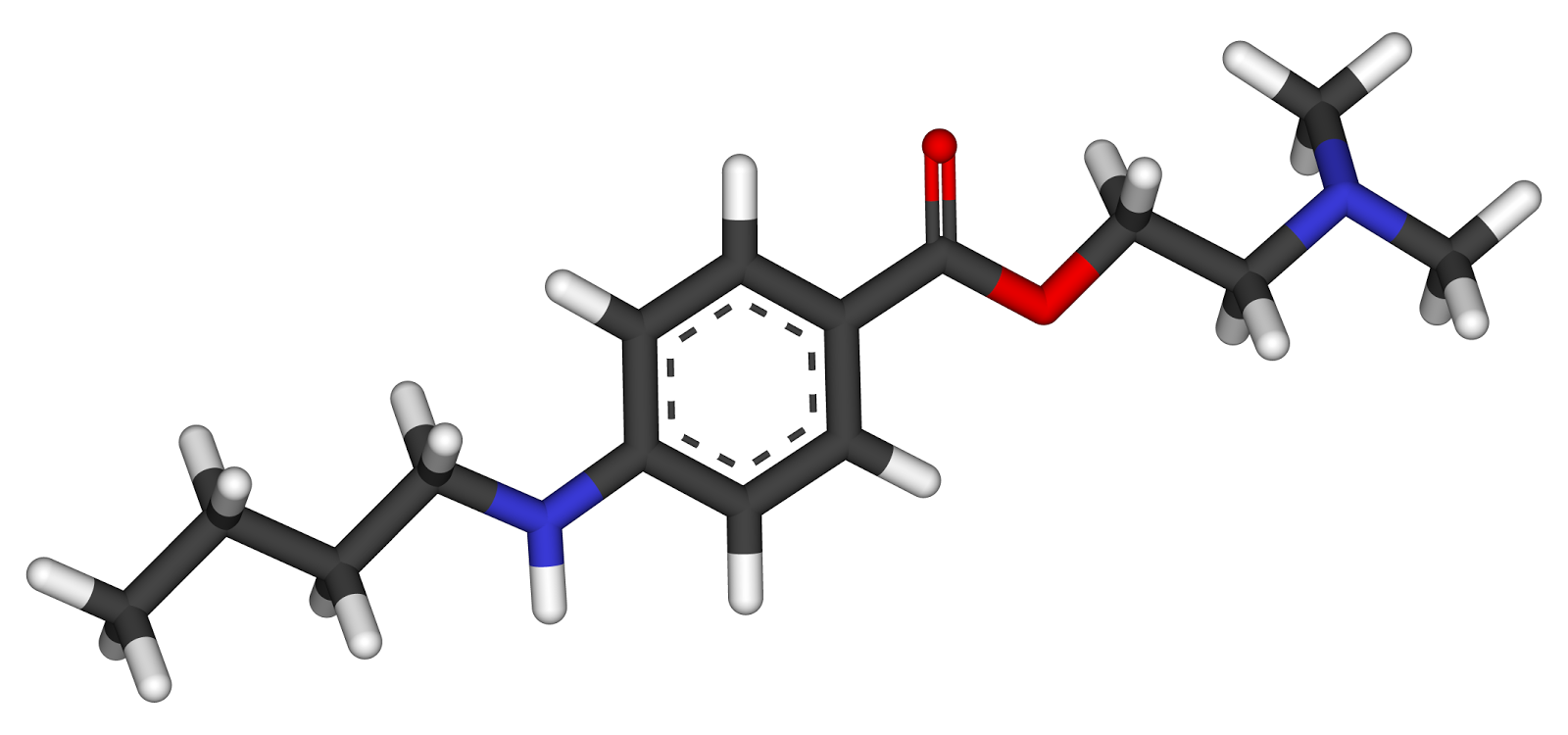
Properties:
- The molecular weight of tetracaine is 264.36 g/mol.
- The molecular formula is C15H24N2O2.
- The systematic name is 2-(dimethylamino)ethyl 4-(butylamino)benzoate.
- The boiling point of tetracaine is between 362.4 degrees Celsius and 416.4 degrees Celsius at the standard 1 ATM.
 tidepressanst that are currently on the market, representing a now aged class of treatments combating depression. Muscarinic, histaminergic and α1-adrenergic receptors are antagonized in the action of classical TCA drugs, leading to anticholinergic (rendering inactive the neurotransmitter acetylcholine), sedative, and cardiovascular effects. In vitro, fluoxetine unites with the aforesaid receptors in the brain tissue with less efficacy than TCA drugs. As identifiable through their names, these TCAs have a three-ring chemical structure. For example,
tidepressanst that are currently on the market, representing a now aged class of treatments combating depression. Muscarinic, histaminergic and α1-adrenergic receptors are antagonized in the action of classical TCA drugs, leading to anticholinergic (rendering inactive the neurotransmitter acetylcholine), sedative, and cardiovascular effects. In vitro, fluoxetine unites with the aforesaid receptors in the brain tissue with less efficacy than TCA drugs. As identifiable through their names, these TCAs have a three-ring chemical structure. For example,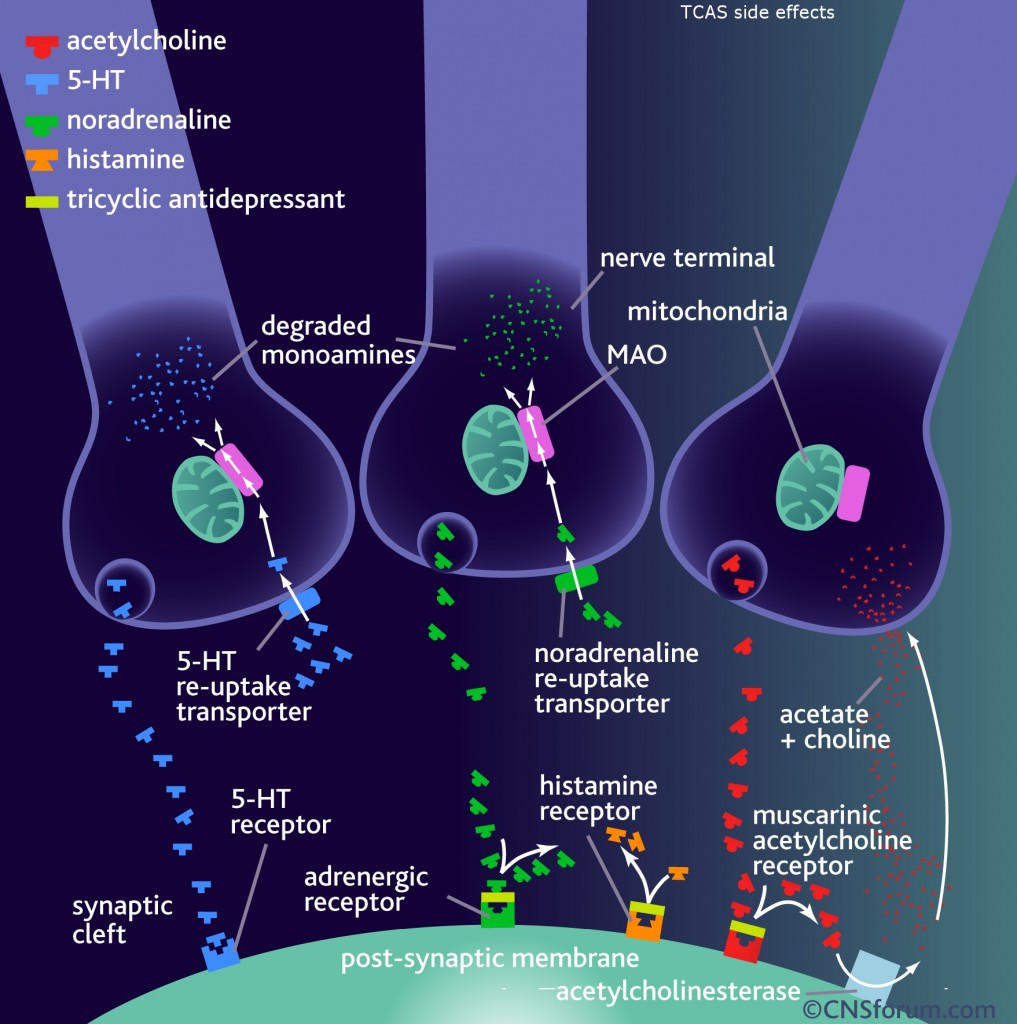 in
in 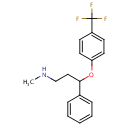 d by the location of an asymmetric carbon atom in the general structure. This is a feature to be noted due to its usages in inorganic, organic, physical, and biological chemistry. It is metabolized by CYP2D6 by the liver, characterized by its slow rate and a long half-life in the confines of the system. Slow aggregation leads to delay in the manifestation of meaningful effect. It is also an agonist for 5HT2C receptors, linking back to the first blog post on beta-agonists.
d by the location of an asymmetric carbon atom in the general structure. This is a feature to be noted due to its usages in inorganic, organic, physical, and biological chemistry. It is metabolized by CYP2D6 by the liver, characterized by its slow rate and a long half-life in the confines of the system. Slow aggregation leads to delay in the manifestation of meaningful effect. It is also an agonist for 5HT2C receptors, linking back to the first blog post on beta-agonists.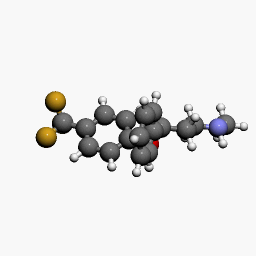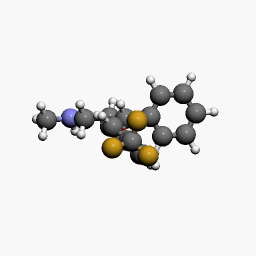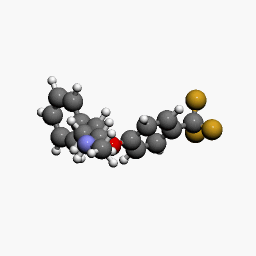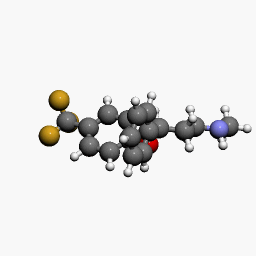



 In both medical practice and research, a placebo is considered an inert substance, or medical dose that is identical in odor, appearance, and taste to an active drug. Clinically, a placebo is defined a substance with no known medical effect that is administered as a control in an experiment to determine the effectiveness of a medical drug. Many times, a placebo simply is a sugar pill. Professionals across several fields are aware that placebos of this definition can cause what is know as the placebo effect. This effect is essentially clinical patients reported the effective treatment of the active drug when a placebo is used. A basic explanation of the idea of a placebo can be found in this video:
In both medical practice and research, a placebo is considered an inert substance, or medical dose that is identical in odor, appearance, and taste to an active drug. Clinically, a placebo is defined a substance with no known medical effect that is administered as a control in an experiment to determine the effectiveness of a medical drug. Many times, a placebo simply is a sugar pill. Professionals across several fields are aware that placebos of this definition can cause what is know as the placebo effect. This effect is essentially clinical patients reported the effective treatment of the active drug when a placebo is used. A basic explanation of the idea of a placebo can be found in this video: be a perspective to the easily tricked human mind, this article from
be a perspective to the easily tricked human mind, this article from 





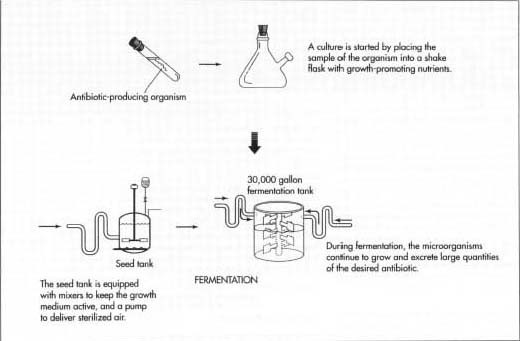
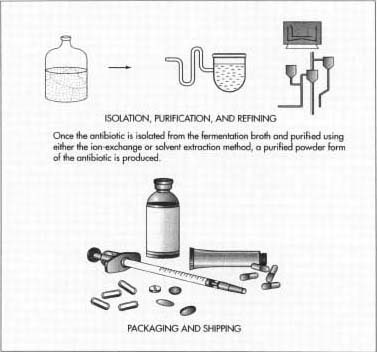


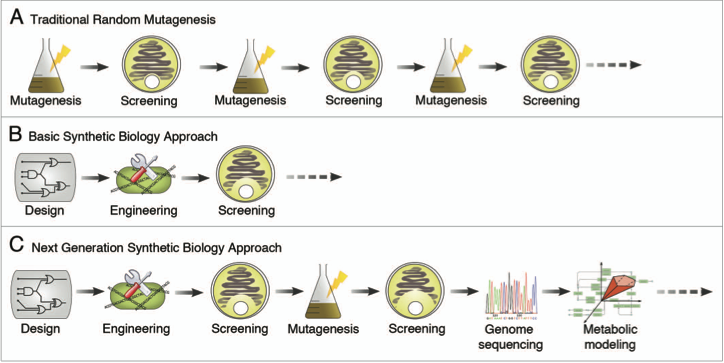 So now that we’ve taken a look at how antibiotics are produced today, let’s see what can be done in the future. This
So now that we’ve taken a look at how antibiotics are produced today, let’s see what can be done in the future. This 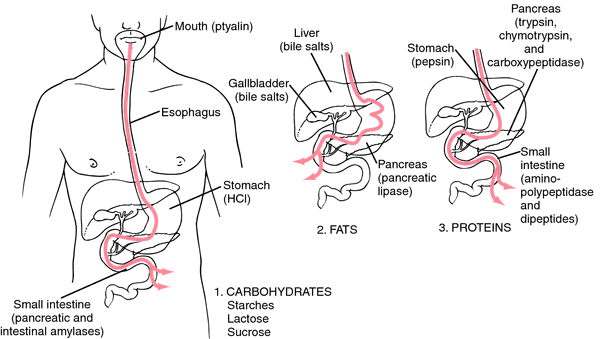
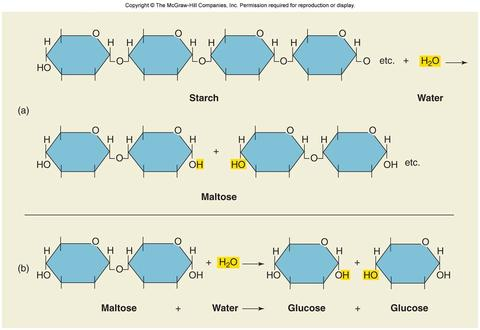






 steel, copper, and nickel. These are just several steps to ensure that the amount of desflurane produced is optimized. Another process that synthesizes desflurane in a relatively inexpensive and environmentally safe manner is reacting
steel, copper, and nickel. These are just several steps to ensure that the amount of desflurane produced is optimized. Another process that synthesizes desflurane in a relatively inexpensive and environmentally safe manner is reacting 
{kind=link}