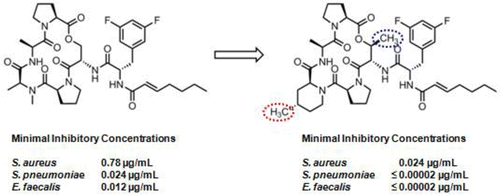
Introduction
In this post we will continue our exploration of the chemistry of antibiotics with a look at drugs that interrupt wall synthesis and how bacteria develop resistance to these drugs.
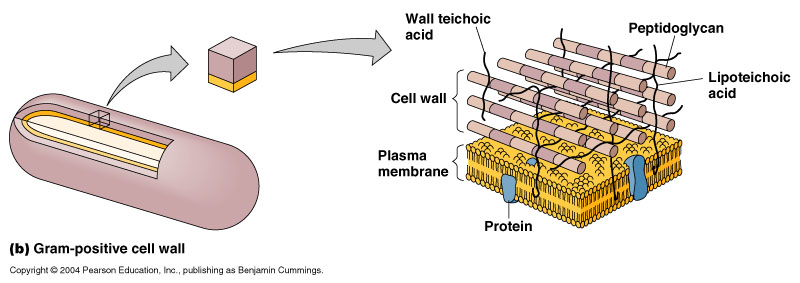
The target of these antibiotics is crucial because it explains how antibiotics can attack bacteria without affecting human cells. Bacterial cells, unlike animal cells, have cell walls. Therefore, a drug that attacks cell walls will not be able to impact human cells. Bacterial cell walls are essentially a peptidoglycan layer that is composed of units of peptides (proteins) and glycans (sugars). This layer is the primary and most important component of the cell wall.
β-lactam Antibiotics
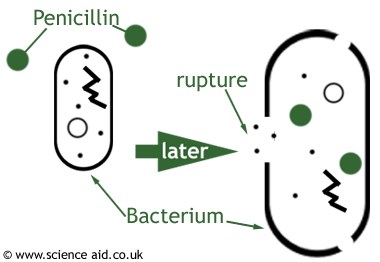
The most common class of antibiotics that interferes with cell wall synthesis are the β-lactam antibiotics, which
include penicillin. These types of antibiotics function by impeding the synthesis of the peptidoglycan of the bacterial cell wall. There are multiple ways to inhibit the synthesis of the peptidoglycan layer but the most common is by means of destroying the enzymes that do so. If the antibiotic does not function by destroying synthetic enzymes, it could also destroy the enzymes that convert the polymers into a layer of the cell wall. β-lactam antibiotics inhibit cell wall synthesis by preventing the assembly of the peptidoglycan layer. This is done when the antibiotic competes with the polymer at the site of binding.
This video gives a general overview of how peptidoglycans are inhibited by the mechanisms of a beta-lactam antibiotic:
The Chemistry of Penicillin


The β-lactam ring in the penicillin reacts with an enzyme that is used for building the cell wall called DD-transpeptidase. Resemblances between a segment of the penicillin structure and the backbone of a peptide chain of the bacterial cell wall have been used to explain the mechanism of beta-lactam antibiotics.
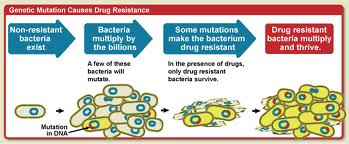
Bacteria Fighting Back
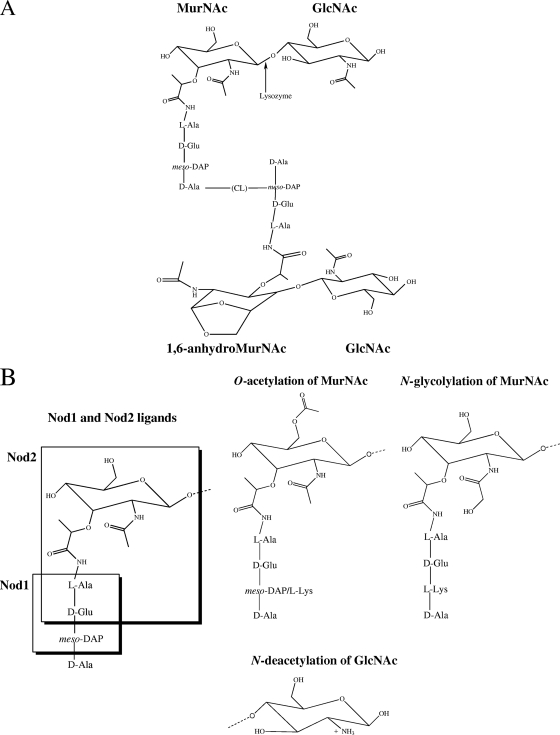
Bacteria have a number of ways to evolve and beat our human antibiotics. One method that bacteria employ in order to fight drugs that interrupt cell wall synthesis, is changing the structure of their cell walls. One instance of this is mutation of pathogenic bacteria. Bacteria that colonize the mucosal pathways, such as nostrils, lips and eyelids, must undergo changes in order to avoid the antimicrobials of the host. In particular, they must avoid lysozyme, an enzyme that destroys the cell walls of bacteria. The activity of lysozyme is similar to β-lactam antibiotics since they both attack the cell walls of bacteria. Since the muramidase activity of lysozyme leads to hydrolysis of the β-1,4 glycosidic bond between the C-1 carbon of N-acetyl muramic acid (MurNAc) and the C-4 carbon of N-acetylglucosamine (GlcNAc), that is the bond that the bacteria have to change in order to survive.Streptococcus pneumoniae and bacillus anthracis, two lysozyme resistant bacteria, have peptidoglycan cell walls that are N-deacetylated, which means that the composition and structure of the sugar component of the cell wall is changed. It was shown that N-acetylating them caused them to be more susceptible to lysozyme. Therefore, it was concluded that N-deacetylating the caused lysozyme resistance. This shows that through changes in structure, bacteria can evade agents that inhibit their cell wall synthesis. When this happens, we too must evolve and change our tactics. This is why new drugs always need to be developed. Bacteria develop resistance, and we must develop new antibiotics.
Want to know more about different classes of antibiotics that inhibit cell wall synthesis? Check out thislink to learn about glycopeptide antibiotics. Of course that’s not enough to satisfy the hunger of your mind, so here is a website that goes further in-depth regarding the processes of cell wall synthesis.

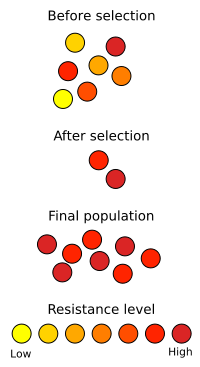


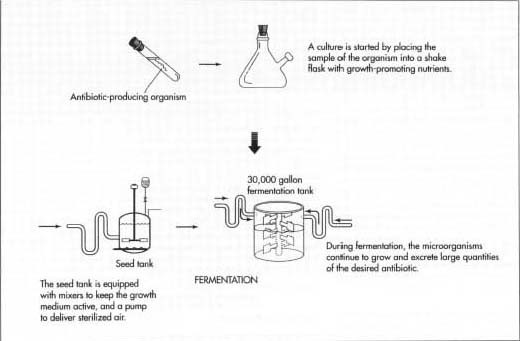
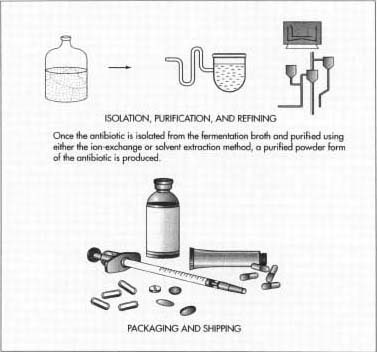


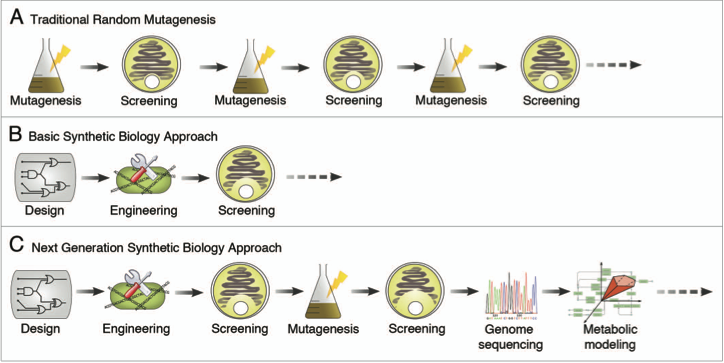 So now that we’ve taken a look at how antibiotics are produced today, let’s see what can be done in the future. This
So now that we’ve taken a look at how antibiotics are produced today, let’s see what can be done in the future. This